F8: esencial para la coagulación y para detener las hemorragias
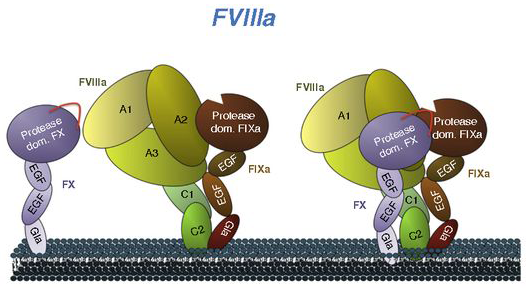
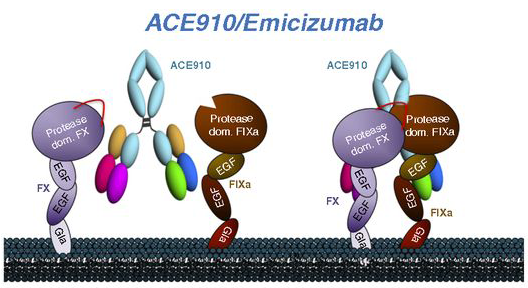
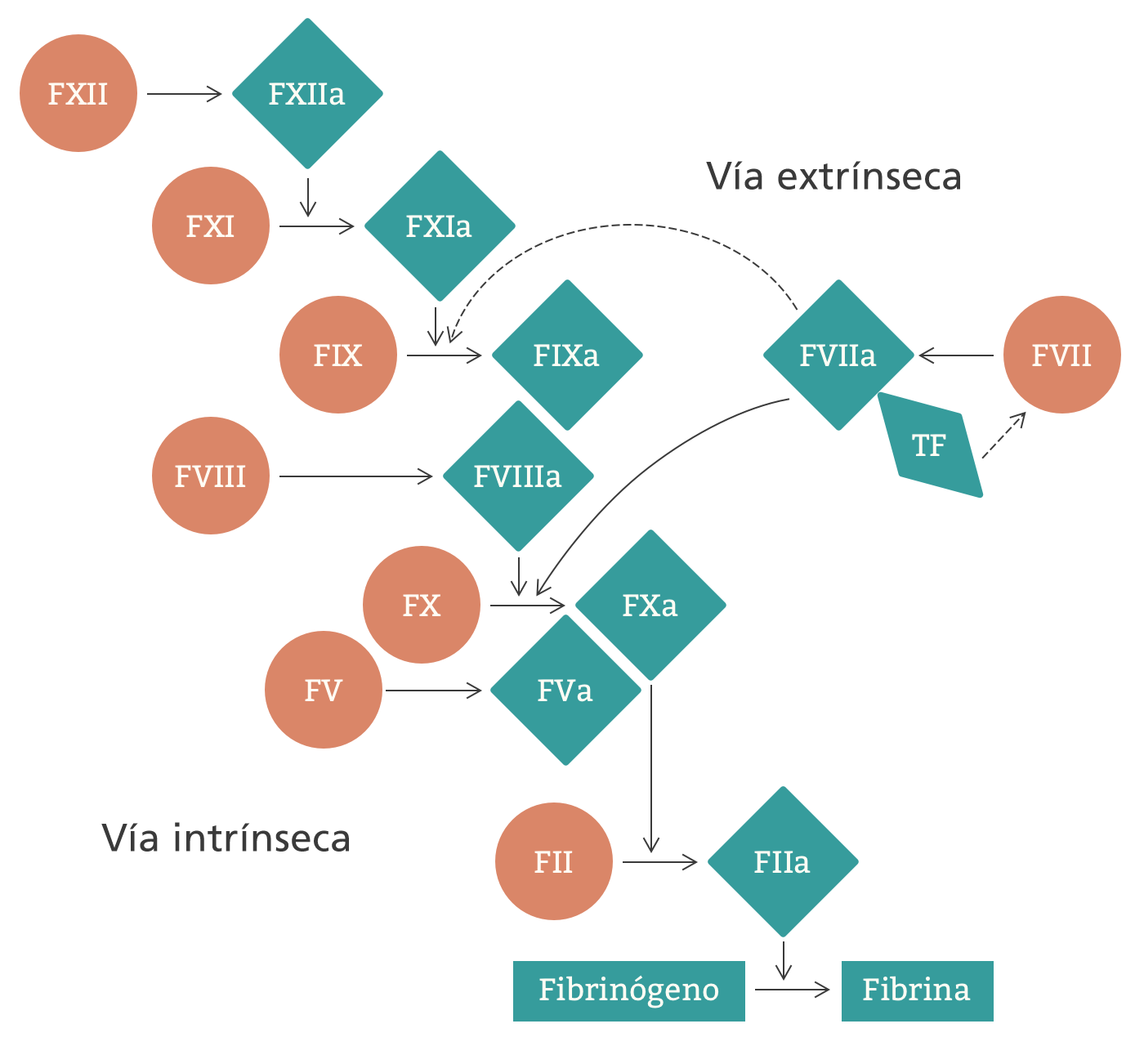
El F8 tiene un rol establecido en la hemostasia (la detención de las hemorragias). Actúa en el lugar de la hemorragia como parte de una cascada de coagulación compleja y estrictamente regulada, que involucra factores, plaquetas, calcio, fosfolípidos y colágenos subendoteliales. El F8 desempeña un papel fundamental, ya que estimula la producción de F10a a través de un proceso de múltiples pasos, lo que culmina en la formación de un coágulo de fibrina estable. Los ciclos de retroalimentación positivos contribuyen a la regulación intrínseca de los componentes de la cascada, y sin una regulación estricta, puede haber caos.
Otras funciones del F8
El F8 desempeña otras funciones en el cuerpo además de ayudar a detener las hemorragias. Estas incluyen el mantenimiento de la salud ósea,2,3 la ayuda para la cicatrización de heridas y la regeneración de tejido,4 la angiogénesis5 (el desarrollo de nuevos vasos sanguíneos) que puede reducir el riesgo de hemorragias en las articulaciones y complicaciones en las articulaciones y, creo yo, posiblemente puede mejorar la integridad endotelial (revestimiento del vaso sanguíneo) para reducir las hemorragias espontáneas. Estas funciones adicionales del F8, junto con su capacidad para detener las hemorragias, pueden verse afectadas negativamente en las personas con deficiencia en el F8.